Application of the Gini correlation coefficient to infer regulatory relationships in transcriptome analysis
Abstract
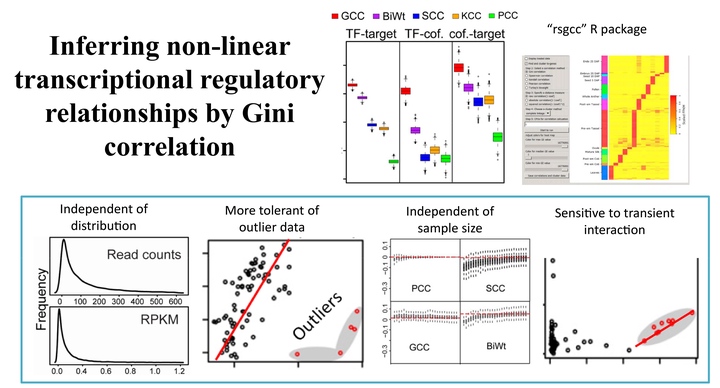
One of the computational challenges in plant systems biology is to accurately infer transcriptional regulation relationships based on correlation analyses of gene expression patterns. Despite several correlation methods that are applied in biology to analyze microarray data, concerns regarding the compatibility of these methods with the gene expression data profiled by high-throughput RNA transcriptome sequencing (RNA-Seq) technology have been raised. These concerns are mainly due to the fact that the distribution of read counts in RNA-Seq experiments is different from that of fluorescence intensities in microarray experiments. Therefore, a comprehensive evaluation of the existing correlation methods and, if necessary, introduction of novel methods into biology is appropriate. In this study, we compared four existing correlation methods used in microarray analysis and one novel method called the Gini correlation coefficient on previously published microarray-based and sequencing-based gene expression data in Arabidopsis (Arabidopsis thaliana) and maize (Zea mays). The comparisons were performed on more than 11,000 regulatory relationships in Arabidopsis, including 8,929 pairs of transcription factors and target genes. Our analyses pinpointed the strengths and weaknesses of each method and indicated that the Gini correlation can compensate for the shortcomings of the Pearson correlation, the Spearman correlation, the Kendall correlation, and the Tukey’s biweight correlation. The Gini correlation method, with the other four evaluated methods in this study, was implemented as an R package named rsgcc that can be utilized as an alternative option for biologists to perform clustering analyses of gene expression patterns or transcriptional network analyses.
Chuang Ma
Principal Investigator, Professor, Doctoral Supervisor
My research interests include artificial intelligence, abiotic stress and plant breeding.